
Catabolism of Phenylalanine and Tyrosine
The aromatic amino acids phenylalanine (Phe, F) and tyrosine (Tyr, Y) share similar structural properties. It is essential to consume phenylalanine-rich foods. However, it is not essential to consume tyrosine-rich foods. Phenylalanine has no other function but to convert to tyrosine after being incorporated into proteins. Tyrosine can, therefore, reduce the demand for phenylalanine in the body. Tyrosine's sparing effect on phenylalanine is referred to as the 'sparing action'.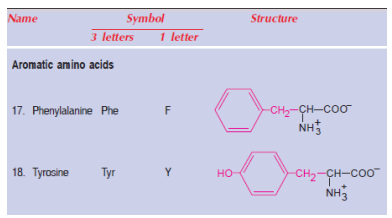
Tyrosine is the main pathway for phenylalanine metabolism. Proteins comprised of tyrosine synthesize epinephrine, norepinephrine, dopamine (catecholamines), thyroid hormones, and the pigment melanin among other compounds.

The degradation process converts the amino acids phenylalanine and tyrosine to metabolites that serve as precursors to glucose synthesis and fat synthesis. The amino acids in these foods are therefore both glucose- and ketogenic.
Synthesis of Tyrosine from Phenylalanine
Tyrosine primarily participates in the degradation of phenylalanine under normal conditions. Phylloquinone (p-hydroxy phenylalanine) is formed by hydroxylation of phenylalanine at para-position by an enzyme called phenylalanine hydroxylase. The reaction is irreversible and requires the coenzyme biopterin, which is required. Biopterin's active metabolite is biopterin H4, a synthetic compound. Tetrahydrobiopterin (H2-biopterin) is oxidized to dihydrobiopterin during the phenylalanine hydroxylase reaction. A dihydrobiopterin reductase based on NADPH regenerated the tetrahydrobiopterin.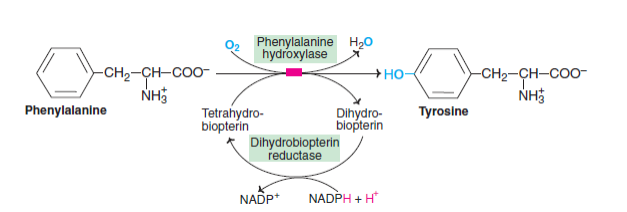
Within the liver is an enzyme known as phenylalanine hydroxylase. Within the liver is an enzyme known as phenylalanine hydroxylase. Tetrahydrobiopterin provides the reducing equivalents to NADPH which, in turn, generates the reducing equivalents. Phenylketonuria (PKU) results from phenylalanine being improperly converted to tyrosine due to a defect in phenylalanine hydroxylase.
Several enzymes are found in the liver, including phenylalanine hydroxylase. Phenylalanine is converted to tyrosine by the addition of an oxygen atom into the para position of the amino acid, while the other oxygen atom is reduced to water. Tetrahydrobiopterin provides the reducing equivalents to NADPH which, in turn, generates the reducing equivalents. phenylketonuria (PKU) is caused by a defective enzyme that converts phenylalanine to tyrosine.
Phenylalanine (Tyrosine) Degradation
Phenylalanine metabolism and tyrosine metabolism are interconnected. Below is a description of the sequence of reactions that takes place in the degradation of these amino acids- Both phenylalanine and tyrosine are degraded primarily in the liver by the same pathway as phenylalanine.
- The p-hydroxyphenylpyruvate is produced by transamination of tyrosine. Tyrosine transaminase catalyzes this reaction (PLP dependent).
- Copper-containing enzyme p-hydroxyphenylpyruvate hydroxylase (or dioxygenase) has a role in cellular sulfur metabolism. p hydroxyphenylpyruvate is decarboxylated and its phenyl ring is hydroxylated to form homogentisate. During this reaction, hydroxyl groups move from para position to meta position, and new groups are introduced at para position. Ascorbic acid is needed for this reaction.
- 4-Melylacetoacetate is formed when homogentisate oxidase (iron metalloprotein) breaks down the benzene ring. To break an aromatic ring, molecular oxygen is required.
- During the isomerization of maleylacetoacetate to 4-fumaryl acetoacetate, maleylacetoacetate isomerase is required.
- Fumarate and acetoacetate are produced when fumaryl acetoacetate is hydrolyzed by fumaryl acetoacetase (fumaryl acetoacetate hydrolase).
Thyroid hormone biosynthesis
Tyrosine residues in thyroglobulin and activated iodine are utilized to synthesize thyroid hormones, thyroxine (tetraiodothyronine) and triiodothyronine. Tyrosine ring iodination leads to the production of mono- and diiodotyrosine, which are then used to synthesize triiodothyronine (T3) and thyroxine (T4). Thyroglobulin breaks down proteolytically to release T3 and T4, which are free hormones.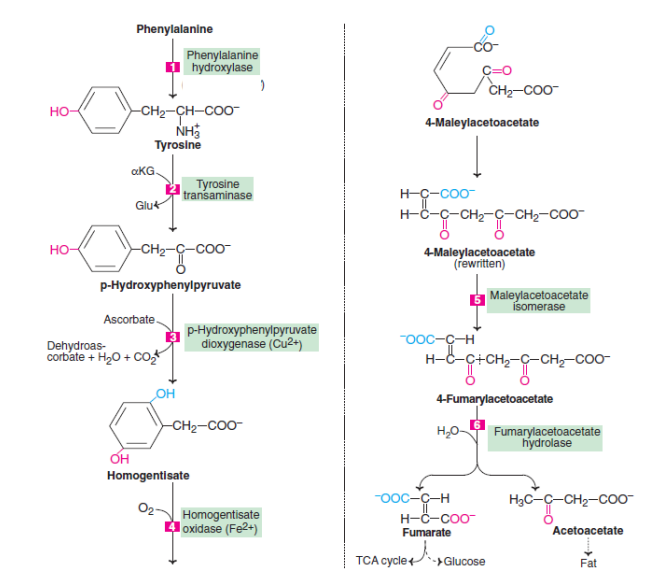

Metabolic disorders
Phenylketonuria
Phenylketonuria (PKU) is an inherited metabolic disorder that results in a high level of the chemical phenylalanine in the blood. Proteins are made in the body using phenylalanine, which is found in most foods. The amino acid phenylalanine can be found in all foods and in a few artificial sweeteners. The body can accumulate toxic levels of phenylalanine without treatment, resulting in mental retardation and other serious complications. When pregnant women consume high levels of phenylalanine, their babies are more likely to suffer from mental retardation, heart problems, and small heads (microcephaly). Phenylalanine is very high in their mother's blood before she gives birth to the babies, so their babies are exposed to it prior to birth.Albinism
A person with albinism is born with the disorder. Both parents with albinism or both parents carrying the albinism gene, have a chance of passing the albinism gene to their child. The cause of albinism is a defect in one of several genes involved in producing and distributing melanin, the pigment responsible for skin, eyes, and hair coloration. Melanin may not be produced or may be produced in very small amounts due to the defect.The gene for albinism is inherited from both parents, so a child must have both parents carry the gene. Parents who carry the albinism gene but don't have symptoms are typically carriers of the condition. Albinism of other types, such as an albinism that affects only the eyes, can be transmitted from one birthing parent to another.
Alkaptonuria
Rarely inherited, alkaptonuria causes kidney problems. The condition is caused by the body not making enough of a compound called homogentisic dioxygenase (HGD). Toxic substances such as homogentisic acid are broken down by this enzyme. Whenever your body does not produce enough homogentisic acid, you build up the acid. In addition to discoloring and brittleness, homogentisic acid can also ruin your bones and cartilage. Your spine and large joints typically suffer from osteoarthritis as a result. As well as turning dark brown or black, alkaptonuria patients' urine also becomes cloudy when exposed to air.Alkaptonuria may also cause the following symptoms:
- There are dark spots on your eye's sclera (white)
- The cartilage in your ears has thickened and darkened
- Discoloration of your skin, especially in the sweat gland area, that is blue speckled
- Sweat of a dark color or stains of sweat
- Earwax that is black in color
- A urinary stone and a prostate stone
- Knee and hip arthritis (especially)
Tyrosinemia
The body does not possess the enzyme it needs for tyrosine metabolism [fumarylacetoacetate hydrolase (FAH)]. The process of metabolism involves breaking down substances, in this case tyrosine, to make energy. Proteins contain the amino acid tyrosine. A person with tyrosinemia breaks down protein in their bodies in an abnormal way, allowing toxic breakdown products of tyrosine to build up. There is progressive liver damage as well as kidney damage. The liver is typically the main site of tyrosine metabolism. A child with tyrosinemia must inherit a mutation of the gene from both parents in order to develop the disease. There is a one in four risk of tyrosinemia in families where both parents have the mutation. There is now a genetic test available that is capable of determining the risk of having a child with tyrosinemia in couples at high risk of carrying the gene. About one out of every 100,000 people is affected by this disease.
Get subject wise printable pdf notesView Here

No comments:
Post a Comment
Please don't spam. Comments having links would not be published.